|
Case Series
Glutaric aciduria type 1: Typical aspects in imaging
1 Department of Radiology Mother and Child, The Children’s Hospital, Mohammed 5 University, Rabat, Morocco
Address correspondence to:
Hajar Zebbakh
16, MOKHTAR Avenue, Kenitra, Radiology Resident, Department of Radiology Mother and Child, The Children’s Hospital, Mohammed 5 University, Rabat,
Morocco
Message to Corresponding Author
Article ID: 100022R02HZ2022
Access full text article on other devices

Access PDF of article on other devices

How to cite this article
Zebbakh H, Diallo I, Lrhorfi N, Alami D, Allali N, Chat L. Glutaric aciduria type 1: Typical aspects in imaging. Edorium J Radiol 2022;8(2):5–9.ABSTRACT
Glutaric aciduria type 1 is an autosomal recessive lysine and tryptophan disorder characterized by glutamic acid and other metabolic by-product accumulation. This disease can be diagnosed in the postnatal period, supported by magnetic resonance imaging (MRI) and confirmed by biochemistry. This article aims to highlight the typical features of this disease in brain MRI which connects frontotemporal atrophy with bilateral and symmetrical signal abnormalities of the brainstem, periventricular white matter, and basal ganglia. As a result, we use two cases to show how this rare disease manifests itself in imaging.
Keywords: Brain magnetic resonance imaging, Glutaric aciduria type 1, Macrocephaly
INTRODUCTION
A metabolic disease with a nervous tropism, glutaric aciduria type 1 (GA1) is an autosomal recessive disorder of tryptophan and lysine that causes glutamic acid and other metabolic by-products to accumulate [1],[2],[3].
It is clinically distinguished by dystonia, macrocephaly, and seizures. The diagnosis is confirmed by neuroimaging, which shows the bat wing sign (frontotemporal atrophy, widening of the Sylvian fissure) with hyperintense lesions of the basal ganglia, along with raised levels of glutaryl derivatives in the blood and urine [4].
CASE REPORT
Case 1
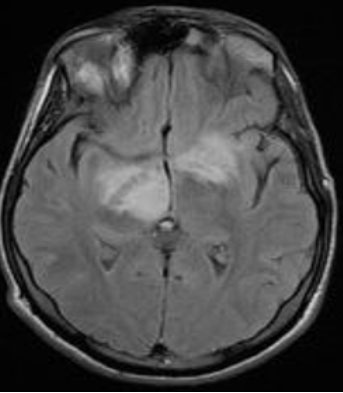
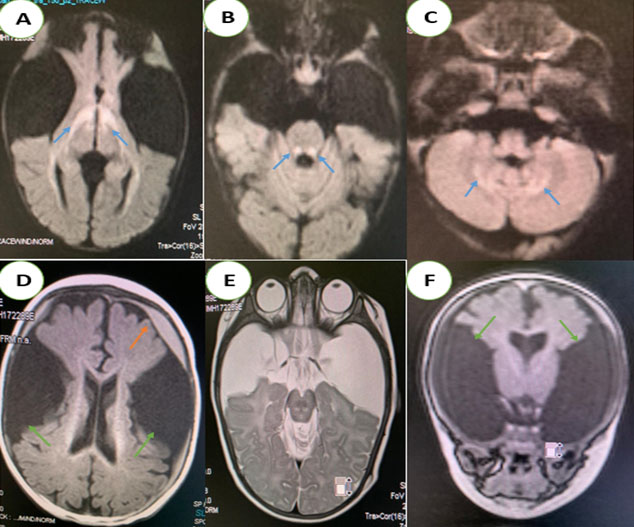
A 10-month-old male infant, the second of two siblings from a non-consanguineous marriage, was referred to pediatric emergencies for dystonia with loss of head support. The history revealed a pregnancy with no particularities, no family context or perinatal asphyxia, while the neurological examination noted axial hypotonia and macrocephaly with retardation in growth. A cerebral MRI was performed (Figure 1).
Case 2
This was a 4-month-old male infant admitted to the pediatric emergency department for apyretic seizures, in whom the history did not reveal any particular context, whereas the physical examination showed a tonic posture of all four limbs with eye revulsion.
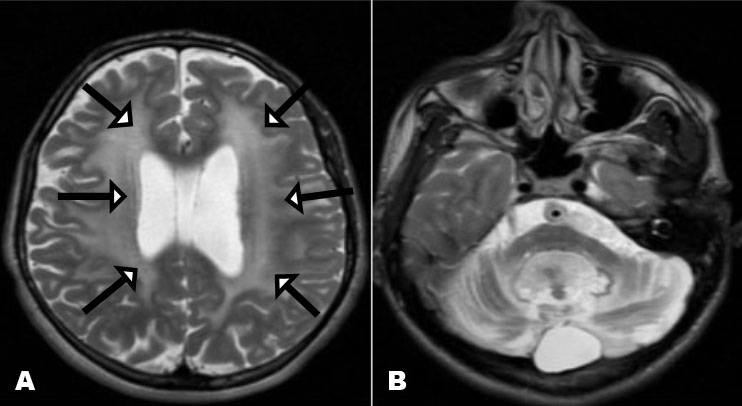
A brain MRI was performed after the resolution of the seizures (Figure 2). The two (2) brain MRIs showed typical abnormalities of this disease:
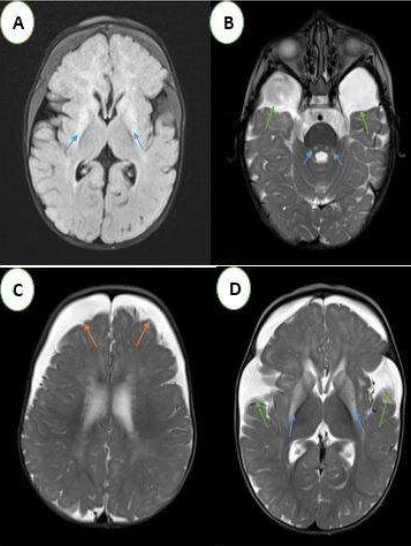
- An enlargement of the temporal fluid spaces and Sylvian valleys.
- Bilateral and symmetrical T2 and Flair hypersignal of the periventricular white matter, basal ganglia, periaqueductal region, and superior cerebellar peduncles, with diffusion restriction.
Frontotemporal subdural hematomas in hyposignal T1, hypersignal T2, without bleeding stigma in T2* and diffusion restriction. In our two patients, an increase in glutaric acid on amino and organic acid chromatography and the detection of a significantly reduced activity of the enzyme glutaryl-CoA dehydrogenase (GCDH) in cultured fibroblasts confirmed the diagnosis. Blood lactic acid levels were elevated.
The infants were treated with riboflavin and carnitine on a diet high in carnitine but low in lysine and tryptophan with good clinical outcomes.
DISCUSSION
An organic acidemia related to a progressive nervous disorder [1],[2],[5], GA1 is an autosomal recessive disease described first in 1975 by Goodman et al. Its overall occurrence is estimated to be 1 in 100,000 newborns [6].
Neurological manifestations differ according to the onset age (from childhood to adulthood).
The symptoms’ severity can range from critical encephalopathy (loss of motor skills, hypotonia, feeding difficulties, and even convulsions) in early-onset variants to more temperate forms in late-onset variants. Early-onset forms are associated with a poor prognosis in contrast to late-onset forms which are low risk [3].
Pathophysiologically, GA1 is distinguished by bilateral striatal neurodegeneration caused by glutaric acid and connected metabolites accumulation, which results in vascular dysfunction with decreased cerebral blood flow as well as impairment of the blood-brain barrier [7].
Primarily, the pathological presentation in GA1 is the loss of gamma-aminobutyric acid (GABA)—which contains neurons along with gliosis—in the basal ganglia [1].
Magnetic resonance imaging revealed bilateral areas of hyperintensity in the caudate and lenticular nuclei within the brains of our two patients, indicating basal ganglia necrosis.
For the evaluation of GA1, brain MRI is the examination of choice. Abnormalities in the bilateral and symmetrical basal ganglia are frequently noted in severely affected children, with distension progressing to atrophy and necrosis [8],[9].
The main characteristic appearance is the widening of the Sylvian fissures in the frontotemporal brain region, resulting in the “bat-wing appearance” [8]. This reversible cerebral atrophy is thought to be caused by apoptosis of immature oligoderms [10].
In severe cases, delayed myelination, macrocephaly, and enlargement of the subarachnoid spaces may also be seen [8],[9]. The MRI signal is characterized by T1 hypointensity, T2 and Flair hyperintensity with diffusion restriction during the acute phase as in our two infants [8],[9].
Subdural hemorrhage may also occur, which is thought to be caused by stretching of the bridging veins in the enlarged meningeal fluid spaces. Its association with frontotemporal hypoplasia is quite common [11].
In GA1, the hematoma might very well form as a result of a mild head shock or appear spontaneously; its regression may be spontaneous. On brain MRI, the subdural hemorrhage is rarely isolated without other signs characteristic of GA1 [11].
When compared with a sex- and age-matched control, spectroscopy may reveal a fall in the N-acetylaspartic acid/creatine ratio; the presence of the lactate spike, which indicates impaired mitochondrial functions, has been rarely reported in this disease [9],[12].
In this case, the diagnosis of type 1 glutamic aciduria was made because of the association of the following radiological signs: frontotemporal atrophy, bilateral and symmetrical signal abnormalities of the basal ganglia, the periventricular white matter and brainstem, as well as the presence of subdural hemorrhage. Very few cases of type 1 glutaric aciduria illustrating all the radiological aspects of this pathology are described in the literature.
These cases have the advantage of being exhaustive because they perfectly illustrate all the characteristics of type 1 glutaric aciduria.
Some patients may exhibit ketosis, mild to moderate metabolic acidosis, hypoglycemia, hyperammonemia, and elevated serum transaminases during acute episodes [2]. None of these metabolic and chemical disturbances were observed in our cases. Elevated glutaric acid concentrations are commonly found in blood, urine, and cerebrospinal fluid (CSF). Urine may also contain 2,3-hydroxyglutaric acid, and plasma amino acid levels seemed to be generally within standard limits [2].
Several GA1 patients, known as high excretors, have elevated amounts of glutaric acid and 3-hydroxyglutaric acid in their urine, whereas others, known as low excretors, possess less apparent organic aciduria [5].
Glutaryl-CoA dehydrogenase enzyme activity should be measured in cultured leukocytes or fibroblasts in any child with progressive dystonia and dyskinesia, as urinary glutaric acid may not be elevated [5].
Increased levels of 3-hydroxyglutaric acid are strongly suggestive of GA1 and the clinician can immediately begin suitable treatment for this disease.
Differential diagnoses arise with other leukodystrophies where macrocephaly is present [13], namely, Canavan disease, Alexander disease, L-2 hydroxyglutaric aciduria, and megalencephalic leukoencephalopathy with subcortical cysts [13].
Bilateral MRI hypersignal of the basal ganglia is seen in some amino acidophiles, mitochondrial diseases, Wilson’s disease, and Zellweger syndrome [13].
Non-accidental injury is a differential diagnosis of glutaric aciduria type I in front of subdural hematoma [13].
Low-protein diets and ones low in tryptophan and lysine are two therapeutic approaches [2],[14].
Riboflavin (as flavin adenine dinucleotide), a cofactor for GCDH, has been suggested to improve residual enzyme activity [1],[2].
L-carnitine has been used to stimulate the formation and excretion of glutaric acid short-chain acylcarnitine derivatives [1],[2],[15],[16].
It acts as an antioxidant or anti-inflammatory agent in this metabolic disease and increases the excretion of toxic metabolites [16]. Glutaric acid also inhibits GABA synthesis.
Gamma-aminobutyric acid analogues like baclofen can be helpful in these situations [2].
Symptomatic treatment, including the management of dystonia, is also indicated.
Finally, regular metabolic assessments by a specialist and a dietician are appropriate.
CONCLUSION
Glutaric aciduria type 1 is a rare disease that should be considered before any dystonia that is accompanied by other neurological symptoms. Brain MRI allows evoking the diagnosis when there is an enlargement of the Sylvian valleys and the peri-cerebral spaces of the temporal poles, with or without striatum involvement. Amino and organic acid chromatography confirms the diagnosis by showing a high level of GA. Drug therapy with riboflavin and carnitine as well as dietary modification can stop the neurological deterioration.
REFERENCES
1.
Lipkin PH, Roe CR, Goodman SI, Batshaw ML. A case of glutaric acidemia type I: Effect of riboflavin and carnitine. J Pediatr 1988;112(1):62–5. [CrossRef]
[Pubmed]

2.
Cornelius LP, Raju V, Julin A. Pediatric glutaric aciduria type 1: 14 cases, diagnosis and management. Ann Indian Acad Neurol 2021;24(1):22–6. [CrossRef]
[Pubmed]
3.
Couce ML, López-Suárez O, Bóveda MD, et al. Glutaric aciduria type I: Outcome of patients with early- versus late-diagnosis. Eur J Paediatr Neurol 2013;17(4):383–9. [CrossRef]
[Pubmed]
4.
Tamhankar PM, Vasudevan L, Kondurkar P, et al. Clinical characteristics, molecular profile, and outcomes in Indian patients with glutaric aciduria type 1. J Pediatr Genet 2021;10(3):213–21. [CrossRef]
[Pubmed]
5.
Goodman SI, Woontner M. An explanation for metabolite excretion in high- and low-excretor patients with glutaric acidemia type 1. Mol Genet Metab 2019;127(4):325–6. [CrossRef]
[Pubmed]
6.
Boy N, Mühlhausen C, Maier EM, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: Second revision. J Inherit Metab Dis 2017;40(1):75–101. [CrossRef]
[Pubmed]
7.
Isasi E, Korte N, Abudara V, Attwell D, Olivera-Bravo S. Glutaric acid affects pericyte contractility and migration: Possible implications for GA-I pathogenesis. Mol Neurobiol 2019;56(11):7694–707. [CrossRef]
[Pubmed]
8.
Mohammad SA, Abdelkhalek HS, Ahmed KA, Zaki OK. Glutaric aciduria type 1: Neuroimaging features with clinical correlation. Pediatr Radiol 2015;45(11):1696–705. [CrossRef]
[Pubmed]
9.
Desai NK, Runge VM, Crisp DE, et al. Magnetic resonance imaging of the brain in glutaric acidemia type I: A review of the literature and a report of four new cases with attention to the basal ganglia and imaging technique. Invest Radiol 2003;38:489–96. [CrossRef]
[Pubmed]
10.
Numata-Uematsu Y, Sakamoto O, Kakisaka Y, et al. Reversible brain atrophy in glutaric aciduria type 1. Brain Dev 2017;39(6):532–5. [CrossRef]
[Pubmed]
11.
Larson A, Goodman S. Glutaric acidemia type 1. GeneReviews®.–University of Washington, Seattle 2019.
12.
Oguz KK, Ozturk A, Cila A. Diffusion-weighted MR imaging and MR spectroscopy in glutaric aciduria type 1. Neuroradiology 2005;47(3):229–34. [CrossRef]
[Pubmed]
13.
Renaud DL. Leukoencephalopathies associated with macrocephaly. Semin Neurol 2012;32(1):34–41. [CrossRef]
[Pubmed]
14.
Gokmen-Ozel H, MacDonald A, Daly A, et al. Dietary practices in glutaric aciduria type 1 over 16 years. J Hum Nutr Diet 2012;25(6):514–9. [CrossRef]
[Pubmed]
15.
Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q. Role of carnitine in disease. Nutr Metab (Lond) 2010;7:30. [CrossRef]
[Pubmed]
16.
Guerreiro G, Faverzani J, Jacques CED, et al. Oxidative damage in glutaric aciduria type I patients and the protective effects of L-carnitine treatment. J Cell Biochem 2018;119(12):10021–32. [CrossRef]
[Pubmed]
SUPPORTING INFORMATION
Author Contributions
Hajar Zebbakh - Conception of the work, Design of the work, Acquisition of data, Analysis of data, Drafting the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ibrahima Diallo - Conception of the work, Design of the work, Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Najlae Lrhorfi - Acquisition of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Dina Alami - Acquisition of data, Drafting the work, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Nazik Allali - Conception of the work, Design of the work, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Latifa Chat - Acquisition of data, Revising the work critically for important intellectual content, Final approval of the version to be published, Agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Guaranter of SubmissionThe corresponding author is the guarantor of submission.
Source of SupportNone
Consent StatementWritten informed consent was obtained from the patient for publication of this article.
Data AvailabilityAll relevant data are within the paper and its Supporting Information files.
Conflict of InterestAuthors declare no conflict of interest.
Copyright© 2022 Hajar Zebbakh et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information.